A retro approach to T cell expansion
By Faith Uwadiae
Edited by William Branchett
Recently we welcomed Professor Phillip Ashton-Rickardt for an insightful lecture and journal club discussion on his latest science paper, describing the identification of a novel protein important for T cell proliferation. (Read the full article here: https://www.sciencemag.org/content/348/6238/995.full)
Effective CD8+ T cells responses are required for robust control of viral infections. However, during chronic infections, including clone 13 lymphocytic choriomenigitis virus (LCMV Cl13), the CD8+ T cell response becomes exhausted resulting in viral persistence. Ashton-Rickardt and colleagues used a forward genetic chemical mutagenesis approach to generate a panel of mouse lines, which they screened for enhanced CD8+ T cell responses during LCMV Cl13 infection. Screening more than 400 lines, the team identified three mutants with the desired phenotype, of which one, named Retro was the focus of this work. Retro mice benefited from increased CD8+ T cells proliferation and cytotoxic activity and almost complete clearance of virus. Interestingly, this improved CD8+ T cell activity in Retro mice was also sufficient to dramatically reduce tumour growth in mice given B16 melanoma cells.
The Retro mouse phenotype mapped to a single Alanine to Glycine mutation at position 1304 of an orphan gene which they named, lymphocyte expansion molecule (LEM). Retro T cells accumulated LEM as the mutation prohibited degradation of its mRNA. Contrastingly LEM heterozygous knockout mice had a 5-fold reduction in their CD8+ T cell numbers and higher LCMV Cl13 viral titre compared to wild type mice. Ectopic overexpression of human LEM resulted in increased human CD8+ T cells. Together this suggested that LEM was a positive regulator of CD8+ T cell proliferation.
Yeast 2 hybrid screening demonstrated LEM interacted with the mitochondrial protein, CR6-interacting factor-1 (CRIF-1), implicating it in oxidative phosphorylation. Indeed Retro mice had an increased rate of oxidative phosphorylation and LEM driven T cell expansion was found to be CRIF-1 dependent. Data from the lab of Erika Pearce (1) has previosuly suggested a link between mitochondrial reactive oxidative species (mROS) generated during oxidative phosphorylation and T cell proliferation. Fascinatingly, Retro mice had increased mROS levels and anti-oxidant administration prevented LEM induced CD8+ T cell expansion. Taken together, this suggests LEM plays an im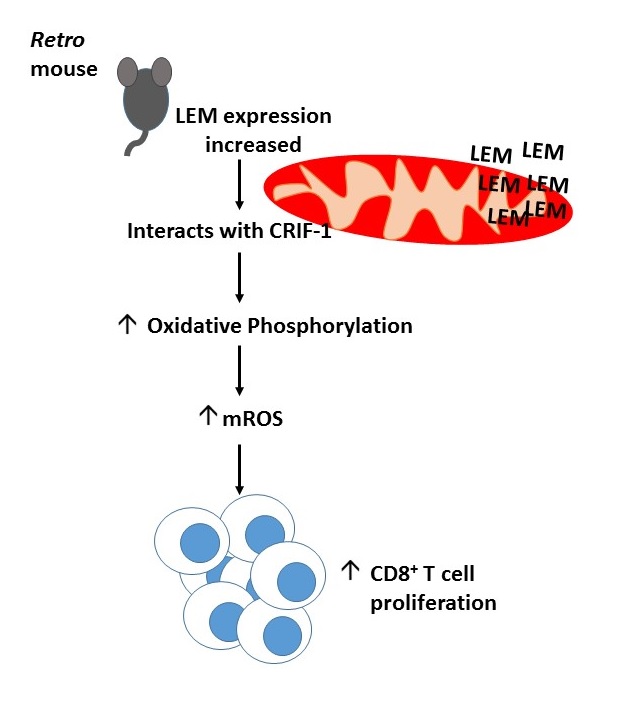 portant role in driving T cell proliferation by regulating mROS mediated oxidative phosphorylation.
portant role in driving T cell proliferation by regulating mROS mediated oxidative phosphorylation.
After 6 long years of elegant molecule biology and dedication Ashton-Rickardt and colleagues have managed to identify a novel molecule with therapeutic potential for the treatment of cancer and chronic viral infection (summarised in the diagram). Much of the journal club discussion with Prof. Ashton-Rickardt focused on the methods of improving cytotoxic immunity by increasing LEM gene expression and the possible caveats of this in the clinic. There was also interest in the localisation of LEM within the mitochondria and the intricacies of mROS mediated T cell expansion. Professor Ashton-Rickardt was a clear advocate for the use of forward genetics to improve our understanding of key biological questions.
The team behind the paper have now formed a company called Immunar and are attempting to patent the therapeutic use of LEM with the hope of reaching the bedside within three years. We look forward to seeing how the story of LEM unfolds.
References
- Van der Windt et al, 2012, Mitochondrial Respiratory Capacity is a critical regulator of CD8+ T cell memory development, Immunity 36 (1):68-78
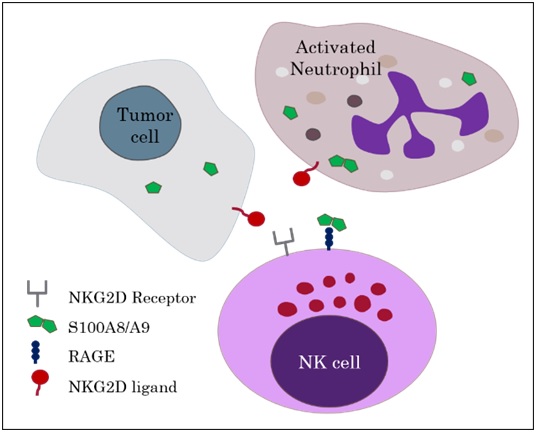 to elucidate these effects but additional binding of NKG2D, an NK cell activating receptor, to an NKG2D-ligand was required. The authors concluded that RAGE/calprotectin has an enhancing or tuning effect on NKG2D induced NK cell activation.
to elucidate these effects but additional binding of NKG2D, an NK cell activating receptor, to an NKG2D-ligand was required. The authors concluded that RAGE/calprotectin has an enhancing or tuning effect on NKG2D induced NK cell activation.