Revolution rather than evolution? A journal club discussion on the use of gene editing based on the Netflix documentary series “Un-natural selection”
Written by Patricia Ogger
Edited by Max Kirtland
For millennia, humans have attempted to control nature to select traits in animals, change the environment, and eliminate infections and disease. In the last decade, the discovery and utilisation of the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) system has provided us with a specific, efficient and cheap method to change nature instantly at the genetic level (see schematic for details – Doudna & Charpentier, 2014, Science).
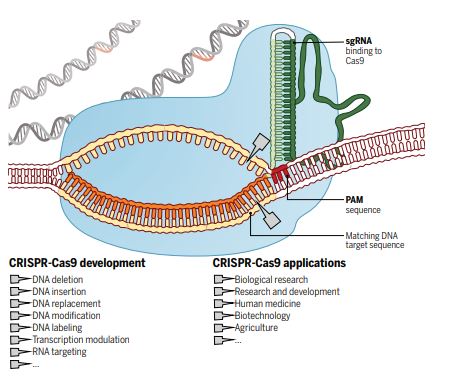
The explosion in the use of CRISPR, and the societal and environmental implications of this and other gene editing techniques were recently covered in the Netflix documentary “Un-natural selection”, by filmmakers Joe Egender and Leeor Kaufman. The four-part documentary series featured scientists, doctors, patients, conservationists and biohackers, showing their attempts and struggles to rewrite the code of life.  It further highlights some examples of applications such as treating rare diseases with specific genetic mutations; inducing gene drive to modify or eliminate wild animal populations to stop transmission of infections such as malaria and Lyme disease or eliminate rats; and the more controversial editing in human babies. The series highlights some critical questions for us as scientists and our society, which we discussed in our journal club:
It further highlights some examples of applications such as treating rare diseases with specific genetic mutations; inducing gene drive to modify or eliminate wild animal populations to stop transmission of infections such as malaria and Lyme disease or eliminate rats; and the more controversial editing in human babies. The series highlights some critical questions for us as scientists and our society, which we discussed in our journal club:
- What could and should gene editing be used for?
- Should these tools be available for everybody to use?
- What is a reasonable price for a live saving/life changing treatment?
- What might be the implications of induced gene drive?
- What ethical concerns does gene editing in human embryos raise?
After the discussion round, we concluded that gene-editing tools should be used for the research and development of new treatments for human diseases, though such applications would need to be tightly regulated. Challenges for this would be to identify diseases that could be treated by modifying a single gene in the patient and to make this affordable. Furthermore, these tools to modify somatic, non-reproductive body cells, should be only available for use in a safe and controlled environment, such as research institutes, to prevent the uncontrolled release of genetically modified organisms, which may have unforeseen consequences on the environment. We agreed that changes to human germline cells, like sperm and egg cells, should be prohibited as these changes would be inheritable and not only affect the individual, but also all their offspring. Therefore, genetically modifying human germline cells could have widespread consequences and would be uncontrollable. In animals however, a method called gene drive is already used to modify germline cells and thereby increase the probability of inheritance of a particular gene. By adding a specific gene to both alleles instead of the usual one, the probability of inheriting that gene increases dramatically, to a point where the whole population will carry that gene. For example, inducing gene drive might be a viable option to reduce transmission of diseases such as Malaria or Lyme disease, but such a drastic change to the mosquito and tick populations might have unforeseen long-term consequences for the ecosystem. It might therefore be safer and more beneficial to investigate other strategies to overcome these diseases in highly affected areas, such as developing better vaccines.
CRISPR may aid the development of complex therapies for rare diseases, as these can be very expensive due to a high chance of failure and small groups of patients, although public health care providers might still cover these expensive treatments. In the UK, the National Institute for Health and Care Excellence (NICE) releases guidelines on usage of therapies covered for certain diseases.
Overall, we concluded that while this documentary did not give a detailed explanation of how CRISPR actually works or a history of its discovery, it is a good foundation for discussion about gene editing and ethical concerns surrounding it. Providing a lay introduction to gene editing as a complex scientific and ethical issue, the series encourages a discussion about tomorrow’s gene editing technology that is ever increasingly on the horizon. We as scientists enjoyed the many different perspectives the series offers, some of which were very different from our own views and experiences.
- “Un-natural selection” https://www.netflix.com/watch/80208833?trackId=200257859
- Doudna, Jennifer A., and Emmanuelle Charpentier. “The new frontier of genome engineering with CRISPR-Cas9.” Science 6213 (2014): 1258096.