Interaction between S100A8/A9 with RAGE triggers a novel mechanism for NK cell activation
Written by Judith Secklehner
Edited by Faith Uwadiae
In our latest Journal Club session we discussed a recent publication in the Journal of Immunology. Narumi et al. investigated the role of S100A8/A9 (or Calprotectin) in a tumor environment and discovered a new role of this protein for NK cell activation via interaction with RAGE (receptor of advanced glycation end product).
The study focused on the heterodimeric protein calprotectin, which is formed of the two Ca2+-binding proteins S100A8 and S100A9. Calprotectin can be found in the cytosol of myeloid cells, but not in lymphocytes. Many intracellular and extracellular proinflammatory functions are attributed to calprotectin, essentially regulation of leukocyte adhesion and migration and promotion of cytokine production. Although it can also act as an oxidant scavenger and possesses antimicrobial activity against bacteria and fungi.
There have been some inconsistent findings regarding the role of calprotectin for tumor promotion or progression. Evidence suggests that elevated levels of calprotectin can amplify inflammation and therefore promote tumor initiation in certain types of cancer. However, some clinical studies indicate that in lung or gastric cancer increased levels of calprotectin were related with better prognosis for patients.
Therefore the aim of this work was to further investigate the in vivo role of S100A8/A9 in cancer. To this end murine pancreatic cancer cell lines were transfected with S100A8/9 cDNA and then inoculated subcutaneously in a syngenic mouse strain. Unexpectedly tumor growth was significantly supressed compared to controls inoculated with non-transfected cancer cells. This effect was also apparent in nude Balb/c mice, which lack thymus derived T-lymphocytes, therefore it was likely that NK cells were responsible for the tumor suppression.
Flow cytometry validated that an increased number of NK cells infiltrated the calprotectin-transfected tumor compared to non-transfected tumors. TLR4 and RAGE, are two pattern recognition receptors known to bind calprotectin. Whilst NK cells do not express TLR4, they were found to be positive for RAGE. To investigate whether RAGE/calprotectin binding was the mediator of the enhanced NK cell activation, antibodies to block RAGE were administered to the tumor-bearing mice. This a
bolished the ameliorating effects of RAGE/calprotectin signalling and increased tumor growth to similar levels observed in control cell lines. However RAGE/calprotectin binding alone did not seem 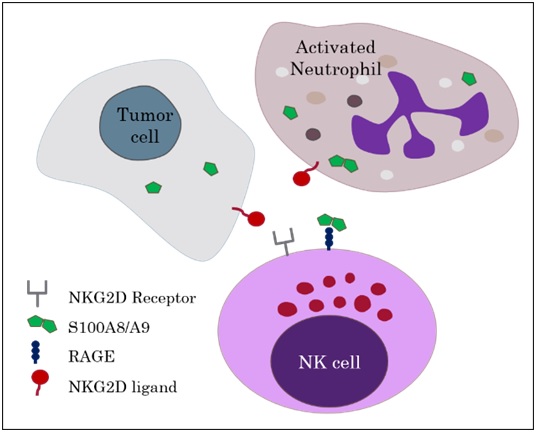 to elucidate these effects but additional binding of NKG2D, an NK cell activating receptor, to an NKG2D-ligand was required. The authors concluded that RAGE/calprotectin has an enhancing or tuning effect on NKG2D induced NK cell activation.
to elucidate these effects but additional binding of NKG2D, an NK cell activating receptor, to an NKG2D-ligand was required. The authors concluded that RAGE/calprotectin has an enhancing or tuning effect on NKG2D induced NK cell activation.
This stimulating paper caused an animated discussion by our Journal Club group focussing on important criteria required for choosing suitable mouse strains and cell lines for a specific model. We were intrigued by this novel finding and are awaiting the future work by Narumi et al, as they intend to investigate the role calprotectin production by neutrophils, for delivering NK cells activating signals to impede tumor progression.
Read the full article at: http://www.jimmunol.org/content/194/11/5539.full